锂离子电池阴离子氧化还原化学的内在科学及实用性讨论
阴离子(X)氧化还原反应的出现,标志着设计正极材料的思路发生了转化型的变革——将近两倍容量的增长。但是,伴随着其发展的是众多的溯源型基础问题以及产业应用上的全能考量。这篇综述里,作者会探讨其中一种可逆的、稳定的阴离子氧化还原行为,并阐释内在机理。进一步地,关于实业应用的限制以及其可行的解决方案也会被条分缕析地罗列以飼读者。另外,作者也专门比对了这类材料与现行主流的高镍系氧化物在市场上正面硬刚的战场空间与胜率。
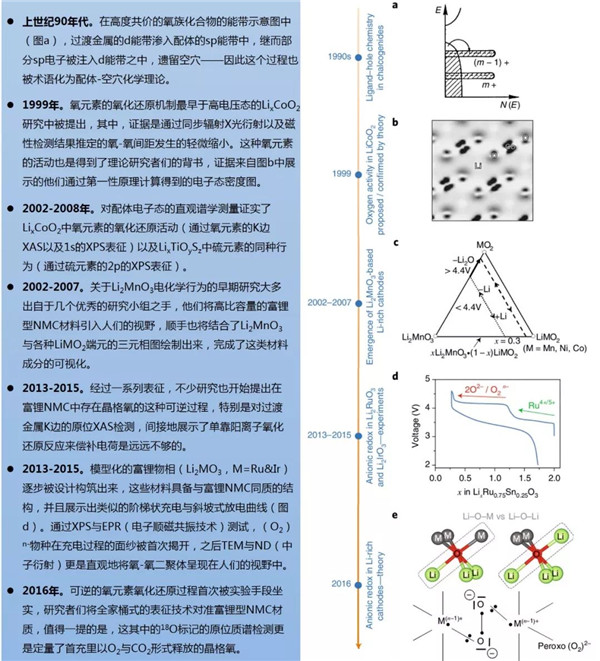
图1阴离子氧化还原化学理论出炉的几个关键节点
阴离子氧化还原化学的内在科学(原理)
在嵌入式复合物的能带结构中,费米能级EF可以与电化学氧化还原(以下简称氧/还)电势关联起来,在这个结构中,费米能级以上的空穴与费米能级以下的电子可以形成一个氧/还电对。含锂过渡金属氧化物的能带结构理论中只考虑到过渡金属d轨道与氧元素的p轨道的重叠效应,这个现象直接导致了成键(M-O)与反键(M-O)*能带的形成,前者具有强配体特征而后者具备金属特性(图1a-c)。(M-O)与(M-O)*能量之差也被称为电荷转移项Δ,其与M和O之间的电负性之差Δχ相关。电荷转移项反映了M-O键的游离/共价特性:例如,将O替换成电负性更低的S之后,Δ降低(离子性更低),并且这个趋势持续着从S直到Te。对于经典的阴极材料来说,它们的氧/还过程只涉及到具有强烈金属特性的(M-O)*能带,因此也常被称作阳离子氧化还原(过程)。
近来,理论学家们通过简单的路易斯描述以及意识到富锂材料能带结构中非键氧态的存在。这个路易斯排布中,O2-含有一个2s以及三个2p的二重态,前者深埋在能级之中不具有氧化还原的活性。相反,高能氧的2p二重态参与到了M-O的成键过程,参与的程度与能带结构有关。不像更高O/M比的富锂Li2MO3材料那样,在经典的层状LiMO2(O/M=2,图1d)中所有的这三个2p轨道都参与到了M-O成键阶段。可以肯定的是,在类Li2MnO3型结构中,氧2p轨道中的一个电子(指向Li1/3M2/3O2层内锂的那一个)的键合比较虚弱,原因是它与锂2s轨道的重叠程度相对较小。因此,它就像一个氧的非键态那样,并固定在稳定了的(M-O)成键能带的上方(图1c)。特别提一下,这个电子也有时被称作氧的2p“孤”或者“未杂化”态,或者“O的孤对电子”,抑或“锂-氧-锂”构型,又或者C2v点群对称中的“b1*态”—— 导致了语义上不必要的困惑。
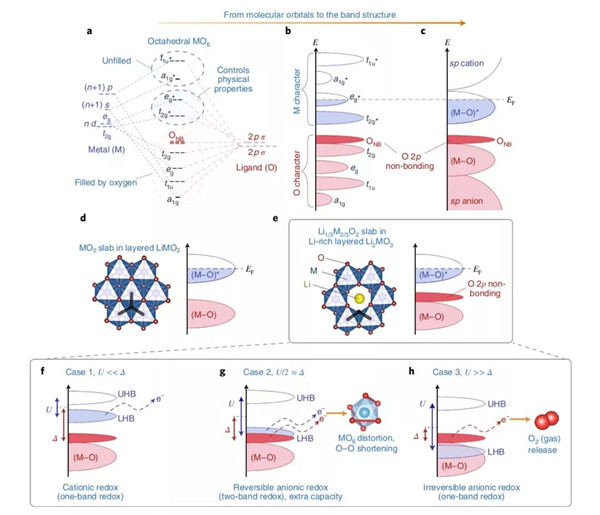
图2. 从分子轨道到能带结构细说M-O配位理论
为什么这个氧的非键态在电化学上如此吸睛?这是因为,与在传统体系中一旦(M-O)*被掏空电子就只能取自于本已稳定的(M-O)成键能带这一变化不同,这个非键态可以作为除了常用(M-O)*能带之外第二个提供额外电子的能带,在不罹患结构失稳的前提下为容量提升添砖加瓦。而能够触发这双能带氧/还过程的关键依赖于(M-O)*反键能带与氧2p非键能带的相对位置。
为了更好的理解能带的占位现象,本文作者首先需要引入d-d库伦相互作用量U,而这并不是一个电池化学家经常引述的明确概念,反而是频频被固体物理学家用来表征d轨道场内电子斥力的术语。这个物理量更接近于描述场内的定域电子而不是动能,它分裂了部分占据的(M-O)*能带(被叫做莫特-哈伯德分裂),形成了空置的上哈伯德能带与满电子的低哈伯德能带(在图2f中分别被叫做UHB与LHB)。更精确的讲,U反比于轨道体积,因此它强相关于参与进来的d金属。所以,它伴随周期表从左至右的轨道收缩而增加,伴随3d至5d过渡金属的轨道膨胀而降低。
LHB位置与氧2p非键能带的相对位置依赖于U对Δ的相对值,这也衍生出三种不同的情形(图2f-h)。首先,对于U<<Δ的情形,其适用于大量的氧化物与氟化物,这些物质具备高度离子化的M-L键(L即配体),电子也取自于满态LHB(图2f)之中,就如经典的单能带阳离子氧化还原情形一样。然后,便是与第一种情况正相反的U>>Δ(图2h),在这种状况下,单能带的氧化还原依旧主导,只不过电子是直接取自于居满态LHB之上氧2p的非键能带。得益于定域氧2p非键态的强化学硬度,这种条件会制造出高度活泼的On-物种,这类物质可能会通过还原消除或者攻击电解液而游离出金属网络。这便导致了常见的Li2MnO3、相关的富锂NMC或者Li4FeTeO6以及Li4FeSbO6阴极中的部分不可逆过程。对于富锂NMC材料来说,如前述报道的附近Mn4+阳离子能在多大程度上稳定On-物种?答案是无法完全稳定,因为随着这类材料爬上首周充电的平台阶段,氧流失的现象始终能被观测到。值得注意的是,这种氧与锂的同时脱出显著地改变了富锂NMC材料的初始能带结构,在之后的过程中它可能会变得更加倾向于可逆的阴离子氧化还原反应以及O-O二聚体的形成。当然,捕捉这种能带结构动力学的变化需要进一步的DFT研究。
最后,U/2≈Δ这种中间情形(图2g),会导致LHB与氧2p非键能带的重叠,这样的条件对于阶段式(Li2-xRuO3)与连续式(Li2-xIrO3)的电化学过程都十分有利,因而可以获得双倍的容量。在这种情况下,电子的转移会导致费米能级的简并,使其失稳。为了规避这种不稳,简并度一般会通过姜-泰勒或者皮尔斯畸变提升,这种效应导致氧框架重组、对称性的降低来缩短某些O-O间距确保稳定化的M-(O2)n-相互作用的发生。这种过氧类O-O二聚体的稳定化作用之前被称作“还原性偶联”,与配位化学颇为相似。以上叙述解释了实验上观察到的Li2IrO3与Li2RuO3中出现的MO6八面体扭曲现象。于是有了一种独特的现象:电子部分地从阴离子的非键能带中脱出,因此把之称为阴离子氧化还原。这与高度脱锂态的LixCoO2全然不同,经Bader电荷计算、XAS与XPS分析表明,高度脱锂LixCoO2中氧是具有氧/还活性的,这是因为其高度的共价性将显著的氧特征传达给了氧/还活性的(M-O)*能带。作者们因此谨慎地反对把这种情况称之为“阴离子氧化还原反应”,因为它基本上也只是一个单能带过程,并不提供额外的容量。
综上,依据这些能带图表可知,要想获得多余的容量,U/2≈Δ是必要的,而这也为材料设计师们打开了大门:通过适当筛选金属-配体的组合来达到Δ与U的微妙平衡。相反地,研究者们应该尽量避免的是不可逆氧流失的情形。理论学家们通过计算3d、4d以及5d金属与氧配位时不同含锂量下氧流失反应的焓变(LixMO3→LixMO3-δ+δ/2 O2)。计算的结果突显了在3d金属配位氧体系中要避免氧流失情形的难度要比4d与5d金属中难得多。考虑到原材料成本之后,这个理论计算的结果尽显悲观,但即便如此,也不应该放弃3d金属配位氧体系,目前已经有不少工艺方面的改进策略在减缓富锂NMC材料的氧流失。
总体上看,无论是强共价性(配位)还是配体中的非键p能带,单打独斗的情况之下都无法满足可逆氧化还原反应进行的条件。例如,只谈共价性,尽管早期对于TiS2的XAS检测表明了S元素具有强力的氧化还原作用,但也仅仅是因为高度共价性的反键(M-O)*态具有明显的配体特征导致的,但这个过程并没有产生额外的容量,至少并没有通过所认知的阴离子氧化还原过程释放容量。同样的,对于氧的非键2p态来说,它们也可以存在于聚阴离子型的化合物中,比如LiFePO4或者LiFeSO4F,但这类离子型化合物因为非键2p态深埋在费米能级之下因而无法催生具备活性的阴离子氧化还原反应,所以它们对外表现为阳离子型的氧化还原机制。
尽管上述场景是建立在氧化物之中,但你也可以将早期的工作推衍至整个氧族化合物之中。比如,在三硫化物TiS3之中,非原位XPS表征的结果清晰明了:锂在电化学回嵌过程中先伴随着S-S二聚体的消失[(S2)2-+2e-→2S2-],然后是Ti4+/3+的还原。简单讲,TiS3中的Ti4+并不能满足S2-态(Ti6+是不可能的),因此引发结构畸变来重新混合空置的硫的3p非键能级以及形成(S2)2-二聚体可以最终稳定TiS2-(S2)2-结构,并且(S2)2-还能如上所述进行电化学还原反应。如果记忆力好一些,你会发现这种阴离子与阴离子的相互作用可以追溯到二聚体之外的一种强共价晶格的物质比如IrTe2[或者说Ir3+(Te3/2-)2],它展示出一种Te-Te亚晶格的聚合来形成所谓的聚CdI2型结构。不过,尽管这种借助全满非键配位能态理论的集大成解释在描述富锂氧化物及相关氧族化合物上令人满意,但在那些新发现的材料上,尤其是针对异常特征它还是会持续受到挑战的。特别指出,同样具备氧非键2p态(指向镁)的贫钠型Na2/3[Mg0.28Mn0.72]O2,之前关于其阴离子氧/还机制的报道结果都基本上复合前述的理论体系范畴。不过,最近在相关报道中,氧活动出现在了具有类似结构但MO2层内并不含碱金属也无碱土金属的化合物之中,这就很让人起兴:这种物质是明显不具有氧的非键2p能态,真相如何还有待进一步探索。当然,除了解释阴离子氧化还原机制之外,这套理论同样也为新型高比容量的材料搜索提供指导意见。
阴离子氧/还机制与富锂阴极材料的实用性(讨论)
在见证了阴离子氧/还反应的高容量潜质如何激发成分与结构多样化的新材料设计之后,作者接下来会评估它们在实际应用中的可操作性,于此你会发现高比容量只不过是几个迫切需求的其中之一。除了具备更高的比容量之外,一个新材料也必须在倍率性能、能量效率与循环稳定性上超越现有的,并且还要在成本与安全上保持竞争力。此外,还需要先进的电化学测试手段(百宝箱3)来专门定睛于与阴离子氧/还型化合物密切相关的特定电化学性质。

图3评估新型阴离子氧/还阴极材料实用性的正确打开方式
以常见的富锂NMC材料开始。尽管经历了数年学术与工业界的努力,这类材料还依旧在等待商业化的春天。从第一周中就可以针砭出实用化的问题,它那特别的两步化第一周充电曲线以一个典型的脱嵌型阳离子氧/还阶段开始,这一过程没毛病,接着就是一个阴离子氧/还阶段(直线区域)并终结于不可逆的产气。除了可以制造出明显破坏电池寿命的寄生反应之外,这种不可逆问题同样会导致全电池的过平衡(更重)。而且,阴离子氧化会永久的改动电化学,并形成S型的充放电曲线。一旦直线区域稳定下来,这种结合了阳离子与阴离子氧/还的活性会提供超高的容量,即便如此,体系仍然会出现大幅度的电压迟滞(>400 mV),从而折损能量效率。这种迟滞源自动力学而不是动力学——也就是说,即便电流接近零也不会消除——并会导致充电vs放电dQ/dV曲线的不对称。它最终会引发路劲依赖,进而使得充电深度(SoC)管理变得十分复杂。由于迟滞减损了能量效率并引发周次的能量损耗(假设以热量形式散发),它还会成为一个成本难题,尤其对诸如电动汽车以及电站等大型应用而言。所以,这个特征(问题)即不容富锂NMC材料忽略,也为过分炒作的那些转化型阳极盖棺定论了。
只有寥寥研究尝试去理解迟滞现象的本质。在这些工作中,电压窗口实验可以展示高电压的阴离子氧化对应于其在低电压的还原情况。核磁共振测试结果辅以相变和格子气模型,认为不可逆阳离子迁移与电压迟滞相关。尤其是在长循环下,这种关联是否暗示因果关系目前尚待商榷。有趣的是,一项最近的研究同样指出了阴离子氧/还反应与阳离子迁移间的相关性,宣称这两种效应是相互耦合的。
不过,人们也必须指导阳离子迁移实际是是阴离子氧化之后结构失稳的结果。顺着这个思路,近来组合光谱技术(HAXPES和XAS)与电化学表征手段展示了阴离子氧化还原与电压迟滞的直接关联。通过关联富锂NMC材料的电化学阻抗与其电荷补偿机制,研究者们还发现与阳离子那快速的氧/还过程相反,阴离子氧/还在高低电压下俱受制于迟缓的动力学过程。电压迟滞再加上动力学迟缓的结果就是,富锂阴极材料的总体极化变大。现今,一个挥之不去的基础问题是,为什么阴离子氧/还反应会与电压迟滞与慢速动力学过程相关,这个问题目前仍需理论上的深入研究。一个合理的假设是能量损耗(因此慢速)的短程原子移动关联到过渡金属持续的、有阴离子氧/还驱动的迁移以及充放电中氧-氧二聚体的持续形成/断裂。因此,氧元素框架的结构弹性可以被认为是一个应对这些问题的关键条件。
在实验方面,尽管历经了大量化学方法的尝试,但富锂NMC中的迟滞问题仍然没有得到解决。有趣的是,在4d金属配位氧型的富锂Li2Ru0.75Sn0.25O3(LRSO)材料中,这个缺陷得到了一定的控制(~200 mV),在这种氧化物中,迟滞现象明显是在阴离子氧化还原反应的高电压阶段被触发的。而且,LRSO中也出现了类似富锂NMC中的dQ/dV曲线不对称现象,但是程度明显要轻。通过对dQ/dV曲线中阳离子-阴离子过程的去耦化,原位XAS检测直观地表明这种不对称现象源于阴离子的氧化还原反应。除了电压迟滞之外,LRSO模型系统也帮助阐释了慢速阴离子动力学过程。综上,在富锂NMC(3d金属型应用体系)与LRSO(4d金属型模型体系)中都有相似的发现,即阴离子氧化还原反应在引发电压迟滞与慢速动力学过充中产生了有害影响,这个发现引人深思,因为其他阴离子氧/还型阴极也出现相似或者更明显的缺陷。举个例子,颇具潜力的低成本、高容量型岩盐Li1.2Mn0.4Ti0.4O2即便在50℃下还是出现了充电与放电电压的巨大差值,不得不让人联想到慢速动力学与电压迟滞。唯一例外于这些缺陷的是Li2IrO3多态物,它们展示出的是叠加型充放电曲线。言而总之,阳离子与阴离子氧化还原反应间的相互作用依赖于化学组成与共价程度,它直接影响了富锂材料中那些及其重要的实用性质,比如动力学与迟滞。
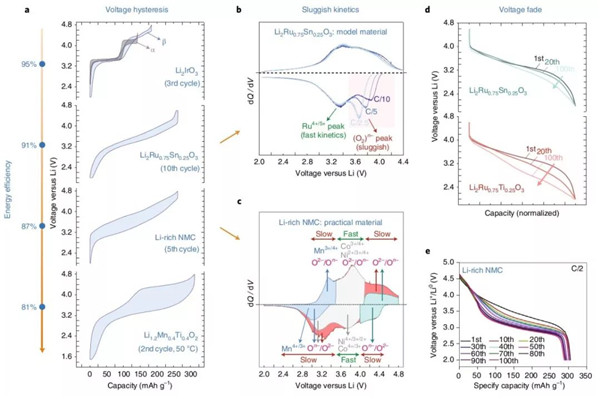
图4富锂材料的现实难题
与这两个相对较新的概念不同,在富锂NMC材料早期研究中便已确认的缺点:电压衰减,已经广受调研,它会逐渐地降低能量输出并使得SoC管理复杂化。现在已经可以明确的是,在高电压下阴离子具有氧化还原活性的富锂NMC材料中,电压衰减明显加剧,这个现象在LRSO模型系统中也得到了进一步的证实。而且,LRSO的研究帮助开发了一种锡稳定型的Li2RuO3改善方案,使得电压衰减在与钛掺杂体系相比之下得到明显的抑制(图4d)。一种合理的解释是,随着长循环的进行,四面体位点逐渐囚困住了小尺寸的钛离子。这种推理同样适用于富锂NMC材料的电压衰减,在高电压下小尺寸的锰离子在氧流失的协助下促进了“层状-尖晶石”的渐变。在其他综述中列出的多种化学方案,从表面改性到化学取代再到电解液添加剂,无一不都是旨在解决富锂NMC材料中的电压衰减。某些方法确实减缓了电压衰减,但完全根除这个缺陷貌似不可能。
最后,以获取更多的阴离子容量高电压充电策略最终会引发氧释放,并恶化材料的稳定性。这种高活性与欠稳定之间的强相关并不陌生,在其他电化学体系中比如水分解催化剂屡见不鲜。在富锂NMC材料首周充电阶段出现的阴离子活化平台是结构失稳的一个早期指标,这个平台反应所释放的晶格氧被认为会持续出现在后续循环中,只不过因为阻氧表面的重构会降低晶格氧释放的速率。阻氧现象也为粒子级设计方案提供了一种动力:保护材料的体相不受氧释放的威胁,这些方案就包括了核壳或者浓度-梯度颗粒的合成。顺便一提,即便是无序岩盐相也无法在长循环下保持稳定。目前,最具潜力的体系是β-Li2IrO3模型,这种可以支撑锂完全脱出的物相突显了三维结构的重要程度,不失为未来开发稳定的阴离子氧/还阴极材料道路上的一盏明灯。
总的来说,富锂材料的实际可行性紧密依靠于阴离子氧/还过程,基于此,改良方案必须直接指向它。这类阴极的缺陷并不会陷它们与万劫不复,问题只是在于如何将合适的阴极匹配进正确的体系与应用。

责任编辑:继电保护
-

权威发布 | 新能源汽车产业顶层设计落地:鼓励“光储充放”,有序推进氢燃料供给体系建设
2020-11-03新能源,汽车,产业,设计 -

中国自主研制的“人造太阳”重力支撑设备正式启运
2020-09-14核聚变,ITER,核电 -

探索 | 既耗能又可供能的数据中心 打造融合型综合能源系统
2020-06-16综合能源服务,新能源消纳,能源互联网
-
新基建助推 数据中心建设将迎爆发期
2020-06-16数据中心,能源互联网,电力新基建 -

泛在电力物联网建设下看电网企业数据变现之路
2019-11-12泛在电力物联网 -

泛在电力物联网建设典型实践案例
2019-10-15泛在电力物联网案例



-
权威发布 | 新能源汽车产业顶层设计落地:鼓励“光储充放”,有序推进氢燃料供给体系建设
2020-11-03新能源,汽车,产业,设计 -
中国自主研制的“人造太阳”重力支撑设备正式启运
2020-09-14核聚变,ITER,核电 -

能源革命和电改政策红利将长期助力储能行业发展
-
探索 | 既耗能又可供能的数据中心 打造融合型综合能源系统
2020-06-16综合能源服务,新能源消纳,能源互联网 -

5G新基建助力智能电网发展
2020-06-125G,智能电网,配电网 -

从智能电网到智能城市